
How FDA Works
A deep dive into America's Favorite Regulatory Body (No Bull Biotech #2)

It’s Game 7 of the 2016 NBA Finals-- the Eastern Conference champion Cleveland Cavaliers are in the middle of a staggering comeback against the defending NBA champion and Western Conference champion Golden State Warriors in a rematch of the 2015 NBA Finals.
The series is tied, 3-3. Rebounding a missed floater by Kyrie Irving with less than two minutes left, Golden State’s Andre Iguodala started the Warriors’ fast break and was about to finish it. He took a bounce pass from Stephen Curry and went up for a layup that would break the 89-89 tie and ignite the Oracle Arena crowd. Except that LeBron James had other ideas, chasing down Iguodala and smacking his attempt against the backboard on the right side of the rim to put the ball back in the Cavaliers’ hands.
The Cavaliers would go on to win the game, clinching the title, and the block would go on to be remembered as the greatest the game has ever seen. We would disagree.
The greatest block of all time is the work FDA does everyday.
This week, we’ll be tearing down how FDA works. Pragmatically, this will mean we’ll be figuring out how we know what we know about drugs, how they get from the bench into humans, and why exactly healthcare is the only industry in love with it’s regulatory body.
Let’s get going.
How did we get here?
If you Google it (and we did)-- FDA is responsible for ensuring the safety, efficacy, human and veterinary drugs, biological products, and medical devices. This is an impossibly broad mandate, so what exactly is going on.
Over the last 150 years, FDA has undergone one of the strangest transformations of any regulatory agency -- growing from a tiny division of the US patent office to the largest consumer protection agency in the world. This history is long, vast, and interesting to people that you hate to be seated next to at a dinner.
Broadly, what we think of as the modern FDA came about as a result of the The Federal Food, Drug, and Cosmetics Act of 1938 requiring all drugs to be approved for safety.
While FDA had an illustrious history chasing down opium and cornmeal counterfeiters before this-- we can really think of the agencies history starting here, and really getting into gear in 1962 when the Kefauver-Harris amendments that added the requirement that drugs be proven “effective” as well as safe. The agency expanded further in the decades following, with the agency reaching its modern state in the 70s with the inclusion of medical devices in its mandate.
There’s been lots of fun detours and filibusters along the way, mostly intended to reduce prices and increase access to a number of essential drugs in rural areas and third world countries.
Much to my mother’s chagrin, turmeric is not in fact an antibiotic, antiviral, antibacterial, anti-infective, cancer cure and cardiovascular health regulator all in one, despite the family anecdotes which would indicate as much-- real drugs are crucial to life in these often medically non-ideal and infrastructure-poor environments.
FDA was also getting tired of the armies of execs showing up in tents and RVs outside of the agency HQ like an LSU tailgate party. The combination of these needs led to the beauty of drug genericization, the Hatch-Waxman act, and downstream, its somewhat unplanned step-sibling acts around biosimilars, “fast-follow” drugs, and more. In this issue of No Bullshit Biotech we’ll dive into what makes a drug a drug, how approvals work, interspersed with the history of FDA and its various evolutions.
What is a drug
In our world, FDA is primarily in the business of regulating drugs. It’s useful here to define a slightly more rigorous definition of drug than “what you find on the floor of Berghain” or “what you take when you twist your ankle for the fifth time that week on the stairs your landlord refuses to fix”.
FDA defines this in five parts:
A substance recognized by an official pharmacopoeia or formulary
A substance intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease
A substance (other than food) intended to affect the structure or any function of the body
A substance intended for use as a component of a medicine but not a device or a component, part, or accessory of a device
Notably this doesn’t include the incredibly grey market world of supplements, herbal products, and vitamins. These are subject to dramatically less scrutiny by FDA’s Center for Drug Evaluation and Research, the ethics of which your authors could not possibly comment on. This is, notably, how all those weird powders twitter users occasionally get prion diseases from get into the country. Your authors could not possibly think that increased regulation would benefit all.
The bounds of exactly what a drug is has blurred over the years, especially with the release and mainstreaming of fortnite, uhhh I mean, digital therapeutics.
Most traditionally, drugs are classified as NCEs or NMEs (often and inaccurately used interchangeably) - throughout history, the majority of NCEs that folks have lining the shelves of their medicine cabinets and pharmacies are traditionally small molecules (groups of elements with a collective molecular weight <500 Daltons). These drugs are often orally available, and tend to cover almost every therapeutic area, from antibiotics to antidepressants to cancer drugs and more.
Next, we’ve got traditional biologics. These drugs are much larger (often between 1 and 200kDa [kilodaltons]), and include peptides like insulin (short strings of amino acids), proteins like interferons (large strings of amino acids) and antibodies like Humira (specialized, large, Y-shaped proteins). There are tons of these on the market, and they’re commonly administered subcutaneously or intravenously.
Over the last few years, we’ve seen the advent of nucleic acid therapeutics, sometimes used as gene therapy (strings of nucleotides expressing or adjusting genetic information, like mRNA), cell therapies (using living cells to cure diseases) and more. All of the aforementioned modalities or classes of drugs can be NCEs or NMEs.
If you hear some folks trading war stories about drugs in a Kendall Square bar, the majority of the time it’s one of these that they’ve taken to market. If you are looking to engage, your authors recommend Miracle of Science in central square, although for some reason they can’t remember their own nights there.
NMEs or new molecular entities are active ingredients in a drug product which contain no active moiety that has been previously approved by the agency - simply, it’s a new drug new drug which has never been approved in any manner (as a different salt, a portion of another active ingredient, etc). They require a New Drug Application, often seen as the most comprehensive and thorough type of approval - often the culprit of the multi-billion dollar drug development cost figures that every AI drug discovery company promises to crush. Congrats $RXRX!
What is approval?
An important tenet to keep in mind when thinking about FDA is that their job is quite different from what most people believe it to be.
They don’t exist to bring new drugs to market or to spur pharmaceutical innovation. They exist to protect patients - they play zone defense instead of crashing the boards.
Simply put, the agency won’t go out of their way to drive companies into the ground by requesting ridiculous amounts of studies or data when existing information is sufficient; they have little to nothing to gain from antagonizing their closest collaborators.
But, where there’s smoke, there’s often fire - a story older than time itself in the pharmaceutical industry. Firms and players are judged by their actions, and reputations are sticky. If data is suspect, if information is inconclusive, and if something is too good to be true (or too bad to be true), red flags will go off and the agency quickly crashes the boards-- requesting more data, often manifested through expensive and more statistically powered (read: more people) studies.
FDA drug approvals are broadly broken into a few different categories. The descriptions below are by no means exhaustive, but are rather meant to give a very high level overview of the different classes of applications that the agency deals with, and the methods by which drugs may be modified, from their core chemistry, to dosage, administration routes, combinations, salts/ester conjugation and more.
Ol Faithful NDAs
NDAs are the bread and butter of pharma. These are for never-before-seen combinations of molecules being tested in people for the first time. They take a lot of work and a lot of money to go all the way, but are also the most defensible and tend to be the most concrete and impactful applications
Eg. Janssen’s Ponesimod (Mar 2021)Add some Salt to it
These are like spicy NDAs - the active ingredient being tested or submitted for approval has already been approved in some manner. The new drug/active ingredient combo is quite similar to the original one, but often has a slight twist; think Lays Salt and Vinegar vs Lays OG. In fact, many times, the modification is as simple as a salt/ester modification (ie. caffeine citrate vs caffeine). These may affect the solubility, bioavailability, or other absorption properties of a given drug, but are often relatively similar to the pure product
Eg. Sanofi’s Clopidogrel Besylate vs Clopidogrel Hydrogen SulfateChopped Cheese
Same same but different - new dosage form applications are used when the active ingredient itself is unaffected, but the manner of getting it into patients is different; ie. going from an IV infusion to a long acting implant, or a subcutaneous injection to a sublingual film. The drug product itself (active ingredient) is the exact same, but the excipients or non-medicinal ingredients are often slightly different.
Eg. Astellas’ oral suspension of Mirabegron (Mybetriq), from an XR tabletDouble Double
When two independently approved drugs (often co-prescribed) love each other very much, they can sometimes perform a transformers/Dragon Ball Z type fusion and produce a combination product lovechild. These are extremely common amongst antivirals and antibiotics where broad coverage is required, along with cardiovascular health medications, consumer health (shoutout Nyquil) and more.
Eg. Axsome’s AXS-05, a proprietary formulation and dose of bupropion and dextromethorphan, both previously approved drugs independently.Reeeeemix
Type 5 applications are a bit of a catch all, and they make up a large portion of genericized applications. Sometimes, it’s not all about the drug ingredient itself - it’s about the vehicle that holds it. New formulation approvals are most often used when the drug product utilises new inactive ingredients that haven’t previously been used in other FDA-approved products, or a different dose/strength of the active ingredient, or a portion of another previously approved combination product, or other bioavailability modifying approaches.
Eg. Almost any generic version of a given drug after patent expiry.Type 6 died in the 90s.
They’re remembered for their debut album, Unknown Pleasures.Trust me it’s chill
Type 7 applications involve an active ingredient which has never previously been approved as part of an FDA sanctioned drug application, but has been marketed in the past - this is most often used for drugs marketed between the 1930s and the 1960s, the types of drugs which everyone knows but weren’t actually “formally” drugs for the longest time.
Eg. think codeine, atropine, and phenobarbital.Something something Hims fixes this
Type 8 applications are used for switching an approved prescription drug to an over the counter product. While some telemedicine startups have more or less turned this into a matter of pressing a couple buttons, it is, in fact, an FDA application route, occasionally seen with simple consumer healthcare products like painkillers, seasonal allergy medication, cough medication and more.Piggyback ride
Type 9/10 applications tag along to an existing new drug application that is already under review (very often a type 1 classic NDA), to streamline the approval of the same drug in a number of different indications in tandem - this type of application is key for “pipeline-in-a-drug” type pharma companies or products, like Alexion’s Soliris, Abbvie’s Humira, Merck’s Keytruda and more.
The vast majority of new drug applications with the FDA fall under categories 1, 3 or 5 - lots of new drugs being developed, tablets being converted to pills or capsules or oral dissolving tabs with minimal changes, and new dose strengths/inactive ingredient combinations being explored.
Gotta Go Fast
Much like a beloved blue hedgehog, FDA recognizes the need to move quickly and to play the objective. Under Scott Gottlieb’s direction, the FDA has worked to appropriately incentivize development of new cures for incurable conditions, first-in-class treatments, and otherwise high-value medications. The FDA makes this happen using the Fast Track Designation, Breakthrough Therapy Designation, Accelerated Approval and Priority Review Designation.
Each of these has a degree of subjectivity in categorizing therapies, but the “rules” for determining a drug’s eligibility follow some common broad strokes:
Fast Track Designation is often used to fulfill an unmet medical need, especially in “serious” conditions, where factors like survival rates, day-to-day functioning, and disease progression may be ameliorated as a result of the drug being evaluated. Examples of such conditions include Alzheimer’s, AIDS, heart failure, cancers, epilepsy, depression, diabetes, and more. Given that these drugs for the most “serious” conditions are often the most impactful, addressing massive markets, and therefore often tend to possess some of the greatest market potential, it’s no surprise that at least 60% of the approved drugs over the past 5 years were fast-tracked. Any drug being developed for a disease with no current therapy is eligible for fast track designation (ie. Graves Disease, Pachyonychia Congenita, etc).
The Fast Track Designation can be implemented at any time in the development process, even before clinical trials start, and allows the company/sponsor to meet with the FDA more frequently to ensure appropriate data is collected, more frequent feedback on trial design and biomarker use etc, along with “Rolling Review”, allowing the sponsor to submit sections of the New Drug Application as data comes in, part by part, as opposed to waiting for everything to be collected and all trials to be completed prior to submitting a full document for review.
Breakthrough Therapy Designation is used for therapies where early clinical data indicates that the drug could significantly outperform the current standard of care on at least one or more clinical significant endpoints. In some cases, that may be the duration of effect (does the drug help for a longer period of time), the magnitude of effect (does it have a greater impact) and potentially the safety (better side effect profile). This can be demonstrated from biomarker data that often correlates to clinically significant improvements, or direct impact on well established clinical endpoints like complete response rates in cancer. Breakthrough Therapy comes with all the perks of Fast Track, along with close guidance from the FDA on development program design, and involvement of senior management on both sides. It’s often implemented prior to Phase III studies, either from strong early stage Phase I data or overwhelmingly positive Phase II results in diseased patients.
Accelerated Approval is granted to therapies for serious conditions with an unmet medical need (similar conditions to Fast Track) based on data from an intermediate or surrogate clinical endpoint as opposed to a known or standard clinical endpoint. Think “tumour size reduction” vs “overall response rate” for cancer drugs - while the former does not imply the presence of the latter, they’re reasonably correlated and one tends to lead to the other. These expected outcomes are tracked in phase IV post-marketing studies once a drug is already on market, to confirm that the intermediate endpoint does in fact lead to the expected final endpoint. These studies can reduce overall time to market, as the sponsors only need to show that these partial endpoints are met, as opposed to waiting for a certain number of patients to survive (or not) etc.
Priority Review Designation is used to drop the standard review time for a New Drug Application from the standard 10 months to 6 months. It’s often used for drugs (or new forms/reformulations of drugs) that are have data indicating an improvement over the standard of care, through increased effectiveness, elimination of treatment-limiting drug reactions, enhanced patient compliance, or safety and efficacy being demonstrated in a new patient group (ie. expanded indications for a known cancer therapy).
All of these represent major process improvements for FDA-- and have all lead to accelerated development, improved economics for drugmakers, and at the end of the day, radically improved outcomes for patients. Even just a few years back, it would be impossible to imagine this level of process improvement in approval pathways-- and marks the start of the modern, responsive, and ultimately more efficient FDA.
Was my summer in Ibiza technically one long clinical trial?
Probably. Enrollment is a lot easier over there. A bigger question that we tend to face is “What exactly is a clinical trial anyway?”
Clinical trials are studies conducted with either healthy volunteers or patients with a given disease, led by a principal investigator who is almost always a medical doctor. They can take place at hospitals, universities, private doctor’s practices, or even community clinics.
Depending on the purpose of the study, patients may be “enrolled” or take part in the study as a result of various physical, biological of chemical eligibility requirements - whether that be age, sex, BMI, blood levels of a certain hormone, presence of a genetic mutation, the list goes on and on. Effective trial design is a massively under-appreciated science, which will be the focus of a whole post in the future. At their core, clinical trials are carried out to understand if a drug is broadly safe and effective, and if there are certain subpopulations where they are especially effective (or dangerous)
These studies are split into different phases, from 0-4.
Phase 0 - Pharmacokinetic and/or pharmacodynamic studies in healthy patients. Small, non-therapeutic doses (as extrapolated from animal models) are conducted in groups of 10-15 or less patients, analyzing the ADME profile (absorption, distribution, metabolism and clearance/excretion) - how the drug breaks down in the body, how it affects various biological processes within the body, and more.
Phase 1 - Safety / dose ranging studies in healthy volunteers without any underlying health conditions. These studies work to identify the maximum safe dose of a drug that may be administered without toxic or problematic side effects. They are often conducted in groups of 20-100 patients, and may sometimes demonstrate early signs of effectiveness (biomarkers, enhanced expression of desired response, etc). About 70% of drugs make it through Phase Is. These often take a few months.
Phase 2a - Dose identification studies in patients with the condition being treated to identify the ideal dose for maximum therapeutic benefit. More isn’t always better for all drugs - a classic example of this principle is the Janus Kinase inhibitors, known for their “bell-curve” like response profiles. Often carried out in 50-300 patients, depending on the indication. About 33% of drugs that made it through Phase Is make it through Phase IIs.
Phase 2b - Preliminary efficacy studies in patients with the condition being treated, using the “best” dose or a range of doses identified from Phase 2a studies, to identify which results in the greatest therapeutic benefit. Some of these may be designed as randomized trials, with placebo vs treatment groups - depends a ton on the indication. The patients are often split into different treatment groups or cohorts, depending on the number of variables being explored (often dose or combination with other drugs). These studies serve to establish initial efficacy or lack thereof for a given drug - does it actually work? Is there any short term toxicity? These studies (combined) can take anywhere between a few months to a couple years.
Phase 3 - Randomized safety and efficacy studies conducted across a range of sites, comparing against placebo and/or standard treatments, in 1000-3000 patients with the condition being treated. These trials are often double blinded to reduce bias, so that both patients and investigators are unaware of what treatment the patient is receiving. Given the larger number of patients, the distribution across sites, and the extended length of most of these studies, these studies help to catch rare and long-term side effects, while eliminating limitations of the Phase I/II trial structures. The larger number of patients, randomization, and distribution of treatments across sites allows the trial sponsors to demonstrate that the drug was safe and effective in a population that’s more representative of the population at large, which might receive such a drug upon approval. If the experimental drug being trialed is found to be more effective than the standard of care and placebos, it often stands a good chance of approval. Phase IIIs are often the final step before a New Drug Application is filed, and approximately 25% of drugs that made it through Phase IIs will proceed through Phase IIIs.
Clinical trials are an essential part of the drug development process.
When run and designed efficiently, they’re a thing of beauty - delivering life-saving treatments to patients who need them most. When sloppily assembled, they’ll lead to more suffering, waste taxpayer money, and set companies up for failure. Note to anyone running or thinking of running a biotech co who is reading this post - don’t skimp on your clinical design team.
Real World Evidence
If there’s any central thrust to this series we’re writing-- it’s telling the story of what an industry can look like when its central regulatory body is effective, transparent, and progressive. I know we’ve harped on it already, but understanding the modernization of FDA under the banner of Real World Evidence is central to understanding everything that will happen in the next decade of healthcare/biotech.
In our intro to this series we write off healthcare directly as useful to our work here. This was true then, but isn’t now. To understand RWE is to understand verticalizing healthcare and biotech into a continuous feedback loop. Welcome to the kwokchain.
Over the last decade, healthcare has undergone a transformation in which quality of care has become determined by value provided to patients and outcomes. The previous healthcare paradigm — physician-centric, infrequent care with minimal external data contributing to decisions — has given way to a patient-driven, continuous care cycle where integrating information from a variety of data sources is critical to clinical decision making. If you come away knowing anything about the last decade of healthcare, this is it.
This transformation necessitated opening up the window to the kinds of data that augment decision making in the clinic. A major driver in validating data collected outside typical clinical settings was the 21st Century Cures Act, which was designed to modernize policies for approving drugs and expanding the use of previously-approved drugs.
A major component of the Cures Act concerned how to leverage data from non-clinical trial sources which describe a patient’s status or the care delivered to them. These data sources are referred to as real world data (RWD). When RWD is processed to generate clinical evidence of the benefits or risks of a medical product, the resulting insight is called real world evidence (RWE).
Passed in 2016, the Cures Act required the FDA to develop guidance for evaluating analyses of RWE with drug companies and implement it within two years. Although the original law was scoped to define how RWE would inform drug approval policy, a side effect was the legitimization of RWD across the drug development and healthcare ecosystems. Today, RWD is used to deliver value not just in the final stages in drug development, but also in drug research & development, clinical trials, marketing, insurance policies, corporate strategy in biopharma, and guiding patient care in clinical and large hospital networks.
This is an unbelievably big deal, and will change medicine as we know it. We will be going deep into this in future entries.
Drugs for All
As Ronnie Coleman once said: “Everybody wanna be a bodybuilder, but don’t nobody wanna lift no heavy ass weights”
The same is true of the pharmaceutical industry - “everybody wanna be a drug hunter but don’t nobody wanna fund no expensive ass efficacy studies or discovery programs.”
When looking at the alignment of incentives across the 4 Ps - patients, payers, pharma and physicians, there are a few simple rules.
Patients want cheap drugs that work well and are easy to use.
Payers want to have access to the largest quantities of necessary drugs at the lowest prices in order to optimize margins when providing for patients.
Pharma wants their drugs to stay on patent and protected to avoid competition to maintain control of pricing and maximize market share.
Physicians want effective, cost effective and plentiful drugs for their patients.
To understand how we make everyone happy with generic drugs, we’ve gotta hop in the DeLorean and rewind to 1984’s Roche vs. Bolar Pharmaceutical Co. case. At the time, if Company A owned the patents on a drug and company B conducted R&D / trials with the intention of marketing an equivalent product after Company A’s patents expired, company B could be found to be infringing on Company A’s patents.
Bolar tried to time its research perfectly so that its product could be launched right as Roche’s expired, but they flew too close to the sun and ended up ultimately losing the patent case. This is now effectively par for the course, but 🅱️olar was a little too far ahead of its time.
This case caused congress to re-examine the regulations around drug genericization; the patent legislature which prevented competitors from working on a reformulation/new version of a given drug during its patent life was effectively granting the patented drug an additional 5-7 years of unaccounted-for exclusivity.
And so, Hatch-Waxman was born - incentivizing both branded and generic firms to develop a more efficient iteration cycle for their products. Noting how long it often took to get a product from inceptions and preclinical studies to final approval, branded companies developing NCEs/new drugs are given 5 years of exclusivity post approval during which (regardless of patent status) generic manufacturers are not able to submit FDA applications for new generic versions. The Act also allows for generic manufacturers to pursue abbreviated New Drug Application pathways, taking only a fraction of the time and costs of the traditional NDA process by relying on the brand manufacturer’s existing clinical data.
As opposed to restarting the drug development/approval process from the ground up, the generic manufacturer must simply demonstrate bioequivalence between their product and the branded reference, which can sometimes be accomplished with a single study. We could go on forever about various clinical endpoints, but the quick and dirty takeaway for bioequivalence is “drug product A is pharmacokinetically and biologically nearly identical to drug product B”, within an acceptable range, with the Cmax (maximum concentration) and AUC (area under the curve, or total amount of drug absorbed by the patient) serving as simple checks.
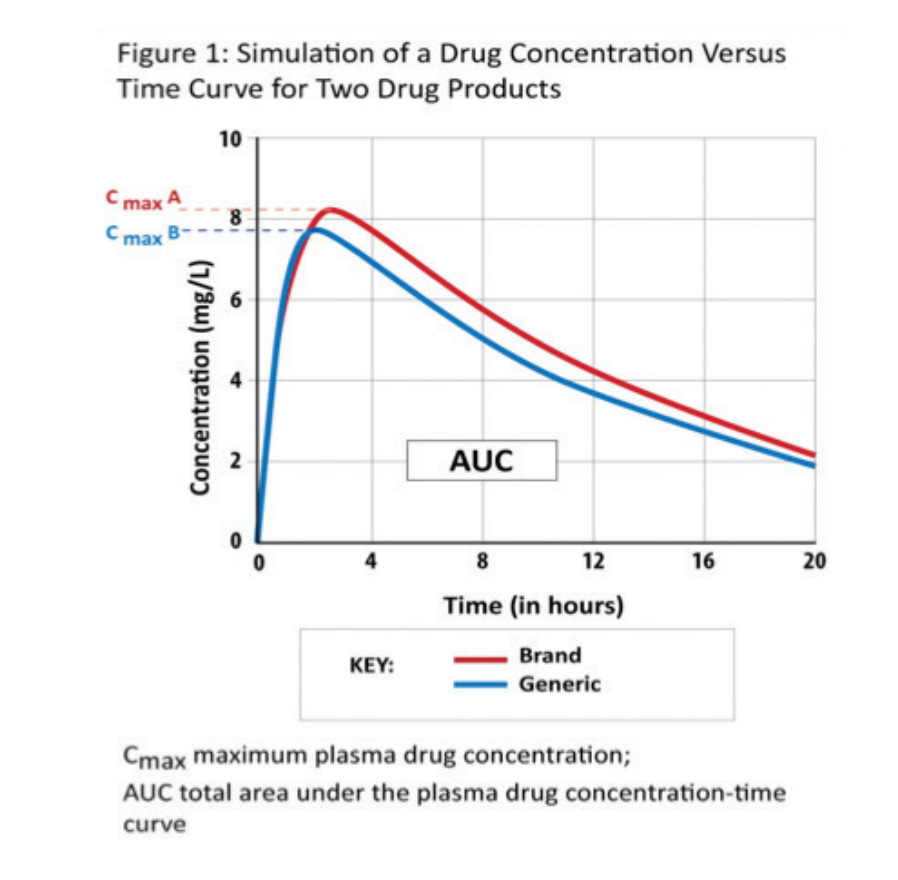
This opened up the floodgates for generic drug applications from household names like Teva, Dr. Reddy’s, Sandoz, and more - allowing generic manufacturers to undercut the prices of brand name drugs while expanding supply and increasing access to essential therapeutics for wider patient populations. Generally, incentives are aligned well for all stakeholders minus the original producer of the brand name drug - but they likely got 10+ years of sales without any competition, so how mad can they really be?
Patients get cheaper medication with identical efficacy to the brand name, physicians have an easier time prescribing medications that they know their patients can afford and access it, and payers have to fork out less $ per pill to their generic pharma suppliers. And for forward-thinking name brand players, they can squeeze extra life out of a drug through a process commonly referred to as lifecycle management, often started anywhere from 5-7 years before the expiry of a composition of matter (read: drug) patent. When life gives you 505(b)2 application eligibility, you make new dosage forms, combination therapies, different formulations, and more, before the generic shops can spit out their own versions.
Abbvie’s long-standing anti-inflammatory blockbuster Humira is the textbook example of effective life-cycle management (and brand marketing!), which we’ll touch on in another post.
So, what’s the end result of genericization?
It’s not all sunshine and rainbows and extreme couponing - there have been horror stories of tainted batches of insulin in foreign manufacturing facilities, as covered in Katherine Eban’s Bottle of Lies. There have been many concerns around counterfeiting, impurities, and more - all of which tend to give generics a bit of a marred reputation / evil stepsister role in the pharmaceutical world. But we firmly believe that the benefits of genericization have outweighed the risks by many magnitudes.
Genericization has led to massive improvements to patient access, quality of life, and pharmaceutical spending. Nearly 90% of prescriptions are now for generic drugs, and they’ve saved the US healthcare system well over $1.5T over the last 10 years alone. Close to 50% of the savings are passed along to the patient directly, resulting in the average patient cutting costs by over $50 per prescription. There are initiatives like Teva’s “penny a pill” program intended to get the average cost of almost any classic drug down to a matter of cents.
Patients all across North America, and perhaps even more importantly, those in less developed countries who cannot afford brand name meds, are getting the essential medications they need at affordable prices. Lives are being saved, diseases are being treated, and incentives are being aligned due to the magic of effective regulation. Pretty crazy to think that it all started with a lineup of tents outside of the FDA office with program leads lining up to get their generic applications in first like a Boxing Day sale.
The generics described above and covered by Hatch-Waxman are most commonly small molecules, but there’s another massive class of therapeutics that the market is moving towards which are also in dire need of affordable and accessible solutions; biologics!
Unlike traditional small molecules which are largely collections of common organic elements, <500 daltons and very often orally available, biologics are large and complex amino acid sequences or proteins made from living cells and often administered subcutaneously or intravenously. Biosimilars are very similar to the original biologic, though they may vary slightly in safety, composition, purity and structure. They demonstrate similar functional, pharmacodynamic and pharmacokinetic properties to the original biologic being replicated.
Biosimilars follow a similarly streamlined application process as generic drugs, allowing these products to be developed for ~10% of the cost of the original drug, and may be brought to market after the original patents for the reference drug have expired. The Biosimilars act of 2010 made all of this possible.
To cap it off
FDA’s prime objective is to keep the public safe. Their secondary objectives are to incentivize the development of medications (novel or otherwise) to broadly improve patient quality of life and accessibility to essential drugs.
The agency has made massive strides towards more progressive and streamlined processes in the last couple of years under Scott Gottlieb’s leadership, nearly singlehandedly taking down Juul, clearing a massive backlog of generic drug applications awaiting approval, and approving a record number of drugs, though industry gossip never ceases to comment on how the approval process is getting tougher. As we venture deeper into therapeutic modalities and potential cures that were once considered pipe dreams, it’s only natural that the agency is closely scrutinizing these potential miracle drugs for any nasty double-edged sword type features.
With the prime objective in mind, they’ve been particularly active recently, halting bluebird bio’s LentiGlobin gene therapy trials after patients being treated for Sickle Cell Disease developed rare blood cancers / disorders. They’re also watching to ensure that the therapies patients are receiving are working as promised, in accordance to the trial design; leading to the initial rejection of BioMarin’s hemophilia A Factor VIII gene therapy as a result of concerns around longer term durability. And of course, we’ve all seen how the agency has intervened at the earliest signs of concern with blood clots and disorders in the roll out of J&J adenovirus based COVID vaccines. We hear about the FDA dunks quite a lot.
In the world of pharmaceuticals, risk is often somewhat proportional to reward until the perfect product is reached.
However, there are a number of wins that you don’t hear about as much. The agency approved the first cell-based gene therapy for multiple myeloma, a targeted exon skipping therapy for Duchenne’s Muscular Dystrophy, the first mRNA vaccines, and countless other potential lifesaving drugs. These new modalities herald the beginning of an era of truly curative medicine - long lasting and potent drugs that can address the root cause of a disease, as opposed to serving as a bandage solution. The future is incredibly bright.
In contrast to a certain caped crusader, the FDA is the hero that we need, not the hero we deserve, with the amount of hate it gets from the general public, capital allocators, and countless other similarly uninformed parties. The agency is here to keep us safe, by whatever means necessary. And sometimes you have to play the role of the villain to actually be the hero.
Feedback, as always, is deeply appreciated. If you’re looking to work in biotech, Science.io and Avro are always hiring. We’ll be back soon to talk about another intractable part of the biotech industry.
would love to see you guys address breakdown of devices side of FDA too!
If I read this before, I would have saved a bundle!
Joe Pulitzer, could not done a better job.
Robert Kremer